您的购物车没有添加商品!
商品分类
质粒载体构建技术综述
质粒载体的构建方法基本上是分子克隆技术,是在分子水平上提供一种纯化和扩增特定DNA片段的方法。通常是用限制性核酸内切酶,DNA连接酶和其他修饰酶的作用,分别对目的基因和载体DNA进行适当切割和修饰,利用体外重组方法将目的基因插入克隆载体,形成重组克隆载体,通过转化与转导的方式,引入适合的寄主体内复制与扩增,然后再从筛选的寄主细胞内分离提纯所需的克隆载体,可以得到插入DNA的许多拷贝,从而获得目的基因的扩增。
文章目录:
1.载体的选择和引物设计
引物设计原则
- 引物设计的目的是将目标片段扩增出来以便与载体连接,所以应在引物的两端加上酶切位点和保护碱基。在选择酶切位点时需注意,应该选择目的基因中没有而载体上有的限制性内切酶,防止目标片段被切不完整,也便于和载体连接。
- 载体构建之后需加入标签以便western blot检测蛋白是否表达。常用的标签有Flag标签、His标签、HA标签和Myc标签。这些标签被表达成氨基酸后,特定的氨基酸序列会与相应抗体结合,在western blot时可被检测。因此,在设计引物时,这些核酸序列应位于起始密码子ATG之后。
- 翻译是从mRNA的5’端开始,而蛋白质合成是从N端到C端,所以若要把标签加在N端,标签所对应的核苷酸序列应位于ATG之后,若是加在C端,则应该将标签所对应的核苷酸序列置于终止密码子TAA之前。
- 对于真核表达载体,蛋白在真核生物中表达时切记加入kozak序列,kozak序列是核糖体在识别mRNA上起始位点时的必须元件,蛋白在翻译表达时是从kozak序列后的第一个ATG开始翻译。并且,kozak序列的存在可以使蛋白的翻译表达效率提高很多倍。
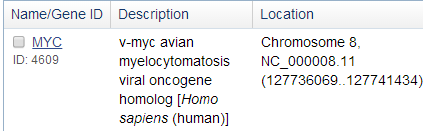
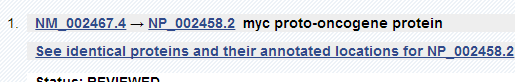
在NCBI中查找MYC的CDS序列



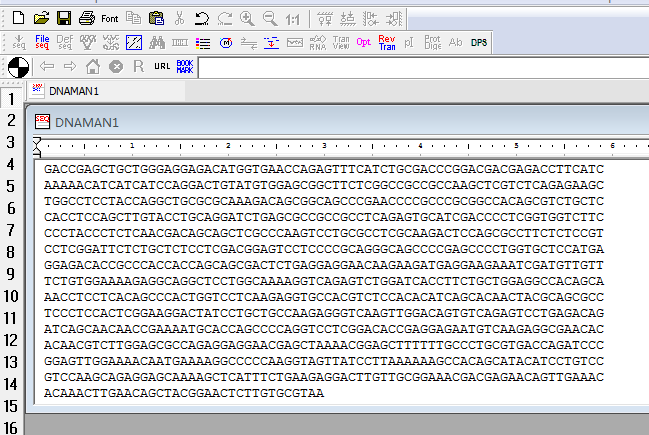
将序列复制粘贴至DNAMAN中分析:
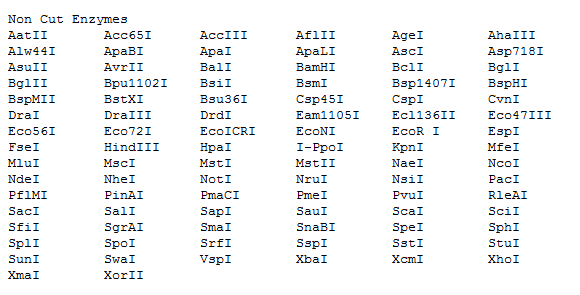
在这些不含有的酶切位点中选择与载体对应的酶切位点,本次举例选择pCDNA3.1
所选择的酶切位点为KpnI和XhoI。
引物设计如下:

如何选择载体
实验室常用的载体有pUC19(扩繁目标片段),pet28a(原核表达载体Kna+),pGEX-4T-1(原核表达载体Amp+),pCI-NEO(真核表达载体Amp+),pCDNA3.1(真核表达载体Amp+),pLKO.1(shRNA构建Amp+)。根据所要构建的质粒目的的差异以及载体和目标片段的酶切位点分析结果来选择载体。详细内容请见文章:如何选择合适的载体
2.目标基因的PCR扩增
PCR技术的基本原理
PCR即聚合酶链式反应,是指在DNA聚合酶的催化下,以母链DNA为模板,以特定引物为延伸起点,通过变性、延伸、复性等步骤,体外复制出与母链模板DNA互补的子链DNA的过程。是一项DNA体外合成放大技术,可用于基因分离克隆,序列分析,基因表达调控,基因多态性研究等许多方面。 PCR反应五要素: 参加PCR反应的物质主要有五种即引物、酶、dNTP、模板和Mg2+
PCR反应的基本步骤:
- 变性:高温使双链DNA解离成单链。(94℃ 30s)
- 退火:低温下引物与模板DNA互补区结合形成杂交分子。(55℃ 30s)
- 延伸:中温延伸。在DNA聚合酶、dNTP、Mg2+存在下, DNA聚合酶催化以引物为起始点的DNA链5’向3’方向的延伸,合成出与模板DNA链互补的DNA子链。(70-72℃ 30-60s)
以上三个步骤为一循环,每一循环的产物均可作为下一循环的模板,经过n次循环后,目的DNA以2n形式增加。
PCR检测
PCR反应扩增出了高的拷贝数,下一步检测就成了关键。荧光素(溴化乙锭,EB)染色琼脂糖凝胶电泳是最最常用的检测手段。电泳法检测特异性是不太高的,因此引物两聚体等非特异性的杂交体很容易引起误判。但因为其简捷易行,成为了主流检测方法。
载体质粒的扩繁
转化(1)取2μl质粒加到1.5mLEP管中,并放置于冰上。
(2)取30μl大肠杆菌DH5α感受态细胞加入质粒中,冰上放置30min。
(3)将水浴锅调至42℃,将上述混合液放在水浴锅中热激1min30s。
(4)取出热激后的混合液,冰上放置2min,加300ul SOC培养基。
(6)取50ul涂布相应抗性的LB固体培养基,放在37℃温箱中过夜培养。
质粒提取(1) 挑取培养基上的单菌落于含有10ml液体LB培养基的50ml培养瓶中37℃过夜培养
(2) 取1-4 ml上述过夜的菌液12000×g离心1min,弃上清
(3) 加250μl Buffer S1悬浮细菌沉淀,悬浮需均匀,不应留有小的菌块。(*确认Buffer S1中已加入RNase A)
(4) 加250μl Buffer S2,温和并充分地上下翻转4-6次混合均匀使菌体充分裂解,直至形成透亮的溶液。此步骤不宜超过5 min。(*Buffer S2使用后立即盖紧瓶盖,以免空气中的CO2中和Buffer的NaOH,降低溶菌效率。避免剧烈摇晃,否则将导致基因组DNA的污染)
(5) 加350μl Buffer S3,温和并充分地上下翻转混合6-8次,12000×g离心10 min。(*避免剧烈摇晃,否则将导致基因组DNA的污染)
(6) 吸取步骤4中的离心上清并转移到制备管(置于2 ml离心管(试剂盒内提供)中),12000×g离心1 min,弃滤液
(7) 将制备管置回离心管,加500μl Buffer W1,12000×g离心1 min,弃滤液
(8) 将制备管置回离心管,加700μl Buffer W2,12000×g离心1 min,弃滤液;以同样的方法再用700μl Buffer W2洗涤一次。弃滤液
(9) 将制备管置回2 ml离心管中,12000×g离心1 min
(10) 将制备管移入新的1.5 ml离心管(试剂盒内提供)中,在制备管膜中央加60-80μl Eluent,室温静置1 min。12000×g心1 min
(11) 取1μl(9)中溶液进行凝胶电泳检测
3.载体和目标片段的限制性酶切
体外构建重组DNA分子,首先要了解目的基因的酶切图谱(如何阅读质粒图谱),选用的限制性内切酶不能目的基因内部有专一的识别位点,即当用一种或两种限制性核酸内切酶切割外源供体DNA时,能得到完整的目的基因。其次,要选择具有相应的单一酶切位点的质粒或者噬菌体等载体分子作为克隆的载体。常用的酶切方法有双酶切法和单酶切法两种。
单酶切法,即只能用一种限制性核酸内切酶切割目的DNA片段,酶切后的片段两端将产生相同的黏性末端或平末端。选择具有相同限制性核酸内切酶识别位点的合适载体,在构建重组分子时,除了形成正常的重组子外,还可能出现目的DNA片段以相反方向插入载体分子中,或目的DNA串联后再插入载体分子中,甚至出现载体分子自连,重新环化的现象,因此重组效率较低。
双酶切法,是构建载体是其中常用的一种方法,使用两种不同的限制性内切酶切割靶基因得到前后都带有粘性末端的靶基因片段,与同样经两种限制性内切酶切割得到的带有相同粘性末端的线性载体在T4DNA连接酶作用下进行连接,从而实现基因克隆。限制性核酸内切酶的类型及特性,按限制酶的组成、与修饰酶活性关系以及切断核酸的情况不同,分为三类:
1.第一类(I型)限制性内切酶:
能识别专一的核苷酸顺序,并在识别点附近的一些核苷酸上切割DNA分子中的双链,但是切割的核苷酸顺序没有专一性,是随机的。这类限制性内切酶在DNA重组技术或基因工程中用处不大,无法用于分析DNA结构或克隆基因。这类酶如EcoB、EcoK等。
2.第二类(II型)限制性内切酶:
能识别专一的核苷酸顺序,并在该顺序内的固定位置上切割双链。这类限制性内切酶的识别和切割的核苷酸都是专一的,因此,这种限制性内切酶是DNA重组技术中最常用的工具酶之一。这种酶识别的专一核苷酸顺序最常见的是4个或6个核苷酸,少数也有识别5个核苷酸以及7个、8个、9个、10个和11个核苷酸的。 II 型限制性内切酶的识别顺序是一个回文对称顺序,即有一个中心对称轴,从这个轴朝二个方向“读”都完全相同。这种酶的切割可以有两种方式:
粘性末端:
交错切割,结果形成两条单链末端,这种末端的核苷酸顺序是互补的,可形成氢键,所以称为粘性末端
如EcoRI的识别顺序为:
5'…… G'AA|TT_C ……3'
3'…… C_TT|AA'G …… 5'
垂直线表示中心对称轴,从两侧“读”核苷酸顺序都是GAATTC或CTTAAG,这就是回文顺序(palindrome)。_和'表示在双链上交错切割的位置,切割后生成
5'G……… 5'.AATTC
3'CTTAA.和3'……...G二个DNA片段,各有一个单链末端,二条单链是互补的,其断裂的磷酸二酯键以及氢键可通过DNA连接酶的作用而“粘合”。
平末端:
II型酶切割方式的另一种是在同一位置上切割双链,产生平头末端。例如EcoRV 的识别位置是:
5'…… GAT'|ATC …… 3'
3'…… CTA'|TAG …… 5'
切割后形成
5'…… GAT ATC …… 3'
3'……CTA和 TAG……5' 二个片段,这种末端同样可以通过DNA连接酶连接起来。
3.第三类( III型)限制性内切酶:
也有专一的识别顺序,但不是对称的回文顺序,在识别顺序旁边几个核苷酸对的固定位置上切割双链。但这几个核苷酸对不是特异性的。因此,这种限制性内切酶切割后产生的一定长度DNA片段,具有各种单链末端。因此不能应用于基因克隆。
4.连接目的基因与载体
在基因工程操作中,外源DNA片段与载体连接的方法主要是通过限制性核酸内切酶和DNA连接酶催化完成的。DNA连接酶催化两双链DNA片段相邻的5’-磷酸和3’-OH间形成磷酸二酯键。在分子克隆中最常用的DNA连接酶是来自T4噬菌体的T4 DNA连接酶,该酶需要ATP作为辅助因子,可以连接黏性末端和平末端。连接反应时,载体DNA和外源DNA的摩尔数之比控制在1:(1~3)之间,可以有效地解决DNA多拷贝插入的现象。反应温度介于酶作用速率和末端结合速率之间,一般是16℃,用常用的连接时间为12-16 h。
5.感受态细胞的制备及质粒转化
转化是指将构建好的重组DNA转入感受态细胞中并进行表达。感受态细胞必须是最容易接受外源DNA片段并实现转化的一种生理状态,其由受体菌的遗传性状所决定,也受菌龄及环境因子的影响。人工转化是通过人为诱导的方法使细胞具有摄取DNA的能力,或人为地将DNA导入细胞内,常用热击法,电穿孔法等。能否实现质粒DNA的转化还与受体细胞的遗传特性有关,所用的受体细胞一般是限制修饰系统的缺陷变异株,即不含限制性内切酶和甲基化酶的突变株。
除自然转化外,细胞经过一些特殊方法处理后,细胞膜的通透性发生变化,也可以成为易于接受并转化外源DNA的感受态细胞。目前常用的感受态细胞制备方法有CaCl2法,简便易行,且其转化效率完全可以满足一般实验要求,制备的感受态细胞暂不用时,可以加入终浓度为15%的无菌甘油,-70℃可保存半年至一年。
经过CaCl2处理的细胞细胞膜通透性增加,成为能允许外源DNA分子进入的感受态细胞。在低温下,将携带有外源DNA片段的载体与感受态细胞混合,经过热击或电穿孔技术,使载体分子进入细胞。进入受体细胞的外源DNA分子通过复制、表达,使受体细胞出现新的遗传性状。将这些转化后的细胞在选择性培养基上培养,即可筛选出重组子。
6.挑取克隆提质粒
平板长出的斑不免存在假阳性,所以在挑菌时应该多挑单克隆,提质粒后进行酶切验证,送阳性克隆测序。挑菌在超净工作台进行,应严格无菌操作,在使用前将所需器材放进超净工作台(除平板)紫外照射20min后方可操作。
7.单克隆的验证及送样测序
重组DNA转化宿主细胞后,并非所有的受体细胞都能被导入重组DNA分子,一般仅有少数重组DNA分子能进入受体细胞,同时也只有极少数的受体细胞在吸纳重组DNA分子之后能良好增殖。并且它们是与其他大量未被转化的受体菌细胞混杂在一起。再者,在这些被转化的受体细胞中,除部分含有我们所期待的重组DNA分子外,另外一些还可能是由于载体自身或一个载体与多个外源DNA片段形成非目标重组DNA分子导入所致。因此必须使用各种筛选及鉴定手段区分转化子与非转化子,并从转化的细胞群体中分离出带有目的基因的重组子。(例如:平板筛选法)
平板筛选法主要用于重组质粒DNA分子的转化子的筛选,而不含重组质粒DNA分子的受体菌则不能存活,α互补筛选法是根据菌落颜色筛选含有充足质粒的转化子。质粒pUC19携带有氨苄青霉素抗性基因(Ampr ),在含有氨苄青霉素平板上筛选转化子。没有导入质粒pUC19的受体细胞,在含有氨苄青霉素的平板上不生长。质粒pUC19进入E.coli DH5α后,通过α-互补作用,形成完整的β-半乳糖苷酶。在麦康凯培养基平板上,转化子利用β-半乳糖苷酶分解培养基中的乳糖产生有机酸,pH降低,培养基中的指示剂变红,转化子的菌落变红。不含质粒的E.coli DH5α,没有β-半乳糖苷酶活性,不能利用培养基中的乳糖产生有机酸,而是利用培养基中的有机碳源,不使培养基pH降低,在不含有氨苄青霉素的麦康凯培养基上形成白色菌落。重组后的载体DNA因为目的基因的插入位点在pUC19乳糖利用基因内部,不能形成α-互补作用,所以也不能利用培养基中的乳糖产生有机酸,在含有氨苄青霉素的麦康凯培养基上形成白色菌落。
挑取阳性克隆提质粒后,应进行验证,对于双酶切连接,采取的是双酶切验证,在验证时体系同酶切连接时类似,但酶量应减少,每个反应体系加0.1ul酶,质粒加入100-200ng即可。对酶切产物进行琼脂糖凝胶电泳,阳性克隆会出现两条带,一条是与载体大小相同,一条与目的片段大小相同。在验证时应做阴性对照,即对载体进行相应的双酶切,其酶切产物只有一条带。
对于单酶切的连接,因为是同样的酶切位点,所以在连接时,可能是ATG在前,也有可能是TAA在前,而我们需要的是ATG在前。应采用PCR的方法验证,在载体上设计一段引物F,与目标片段的R引物一起扩增,只有插入顺序正确的质粒才能扩增出目的片段,错误的无法扩增。
最后将验证后确定正确的阳性克隆质粒送样测序。